
Approval Is a Manufacturing Problem
The FDA released 14 new Complete Response Letters this month, its first batch since it paused publication in April. We read all 14. The pattern matches the previous 291: the clinical data mostly holds up, and the manufacturing is where applications stop.

Byron Fitzgerald
Founder, ProGen Search
Every few months the argument resurfaces that the FDA has become unpredictable. Goalposts move, sponsors get blindsided, and a promising drug is turned away for reasons nobody saw coming. It is a comfortable story, because it puts the failure outside the building.
The letters tell a steadier one. A Complete Response Letter, or CRL, is the FDA's formal notice that it will not approve an application in its present form. Since 2024 the agency has been publishing them: first a set of 202, then a further 89. This month it added 14 more. Read end to end, they are far more consistent than the current mood suggests, and they point somewhere specific.
We turned that pattern into a free tool, the FDA Filing Diagnostic, so you can pressure-test where your own filing is most exposed before the agency does.

What the Letters Actually Say
Of the 14 new letters:
- 10 cite a manufacturing-site inspection: an FDA Form 483, a pre-approval inspection the site could not clear, or a facility assessment that turned up deficiencies.
- 13 carry a manufacturing or product-quality deficiency of some kind, whether tied to an inspection or found on review of the chemistry, manufacturing and controls (CMC) data.
- 3 touch the clinical data at all. Every one of those three also carries a manufacturing or quality problem alongside it.
- 1 has no manufacturing element whatsoever. More on that one shortly.
The categories overlap, because a single letter usually raises several issues at once. That overlap is the point. Manufacturing and quality are not one failure mode among many in this batch. They are the through-line.
None of this is new. The same emphasis ran through the original 202 and the 89 that followed, and an independent read of these same 14 by Citeline reached the same place: only a handful cited clinical, clinical-pharmacology or statistical problems. Three separate batches, one story.
A Short Primer on What Stops a Drug Here
The distinctions matter, because “manufacturing problem” is not a single thing.
A Form 483 is the list of observations an FDA investigator hands a site at the close of an inspection. It is not the CRL itself, but it often sits underneath one. If the findings are serious and unresolved, the application cannot proceed until the site responds to the agency's satisfaction.
A pre-approval inspection (PAI) is the FDA's check of the specific facility named in an application before it will approve that product. For biologics the equivalent is a pre-license inspection (PLI). Increasingly the agency also runs a Remote Regulatory Assessment, a records-based review that can surface deficiencies without an investigator on site, and can still require an on-site inspection afterwards.
Then there is the CMC package itself: the evidence that the product can be made to specification, batch after batch, and stay stable on the shelf. Deficiencies here, in packaging, leachables, stability or process validation, are found on paper, without anyone visiting a plant. It is the less-watched half of CMC risk, and one we have written about before, where programmes come apart in the lab rather than the reactor.
An application can fall at any of these. In this batch, most fell at more than one.
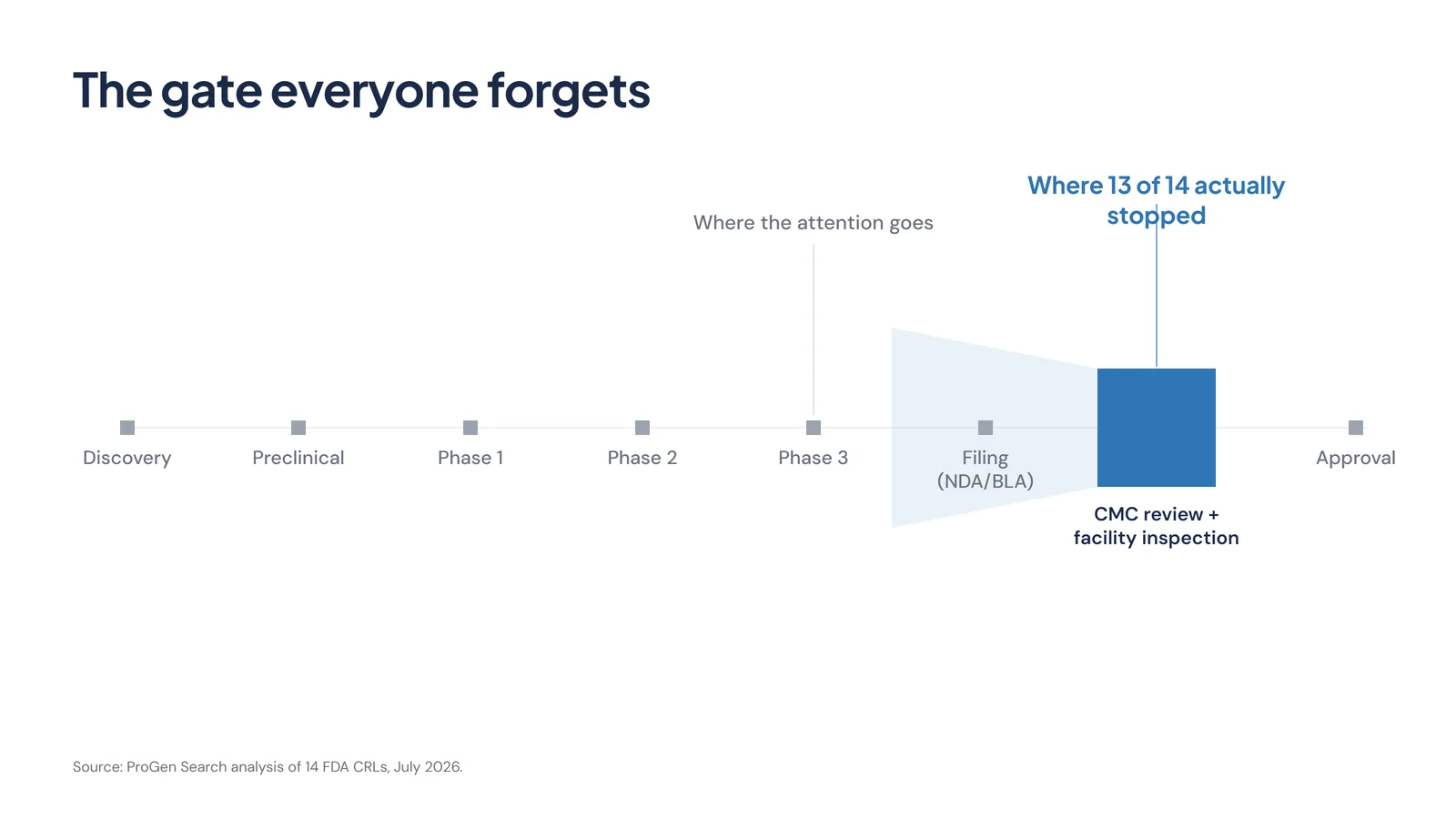
The Gate Everyone Forgets
Picture the path a drug takes to market. Discovery, preclinical work, three phases of clinical trials, then the filing, then review, then approval. Almost all of the attention, the board updates, the analyst notes, the share-price moves, clusters around one point on that line: the Phase 3 readout.
The readout matters. It is necessary. It is also not where these applications stopped. They stopped later, at the manufacturing and inspection gate that sits between a positive filing and an actual approval. That gate draws a fraction of the attention the clinical data gets, and it turns out to be where the queue forms.
That is the asymmetry in how most programmes are run. Clinical risk is watched obsessively. Manufacturing-readiness risk is assumed to be handled, right up to the point where a 483 makes it the only thing that matters.
Two Worth Naming
Two of the 14 are already public, disclosed by the companies themselves, so they are worth looking at directly.
Hengrui and Elevar's liver-cancer combination was turned away for the third time. This round the problem was manufacturing rather than efficacy: the letter cites a CGMP inspection and a Form 483, with further site inspections still required before approval. A combination that clears its clinical bar can still be held by the state of the facilities behind it.
Grace Therapeutics' injectable nimodipine drew a letter centred on product quality, alongside a facility inspection. The company has said it intends to resubmit. That is the texture worth holding on to, because it corrects the instinct to read a CRL as a death sentence. It is not one. It is a delay, and usually a fixable one.
The one letter with no manufacturing element belongs to that same liver-cancer combination. Its second component was refused for a single reason: its partner drug had just been turned down, and the two are only cleared together. Even the exception traces back to a manufacturing inspection on the other half of the file.
None of this is written to embarrass anyone. These are public regulatory records, and in most cases the findings were known to the company long before the letter arrived.
The Regulator Isn't the Story
Which is the part the “erratic FDA” reading misses. Work through the letters and most of these companies had been told. A 483 had already been issued. Deficiencies had already been conveyed. The CRL did not spring an ambush. It made official a gap the site had been carrying for months, between what the facility could actually stand behind and what got filed anyway.
Some of the frustration with the agency is fair. Timelines shift, and reviewers do change their minds. But a batch of letters dominated by inspection findings and CMC data is not the signature of a regulator inventing new reasons to say no. It is the signature of applications reaching the manufacturing gate before they were ready for it.
The molecule is rarely the thing standing between a company and approval. The factory, and the team running it, usually is.
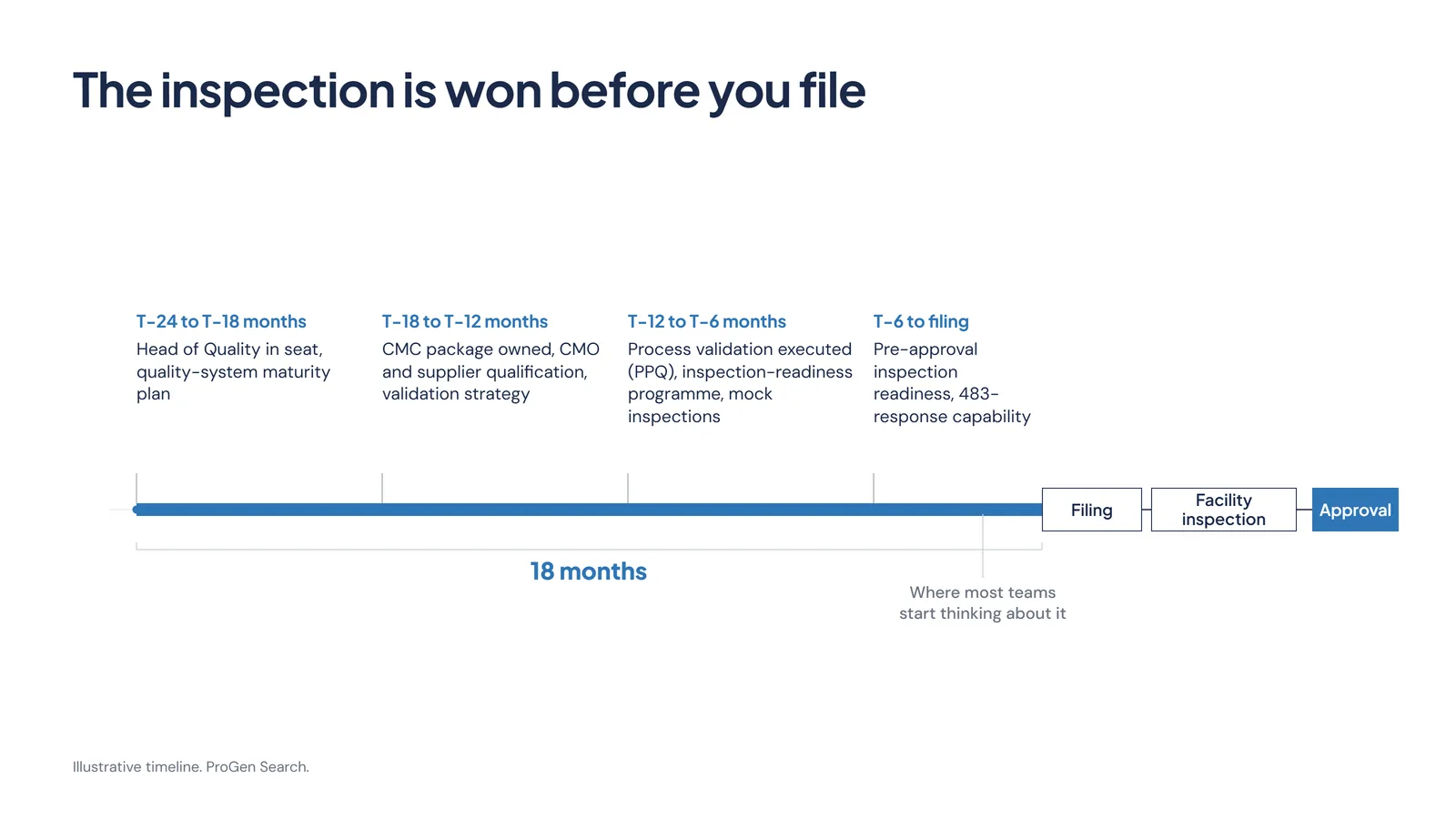
Readiness Is a Hiring Decision
If the gate is manufacturing readiness, then the work that decides the outcome happens long before the filing, and most of it is not scientific. It is operational, and it is organisational.
Process validation closed before submission rather than after. The pre-approval inspection planned for and rehearsed, on the company's terms rather than the investigator's. A CMC package built and owned by people who have been through an FDA inspection before and know what an investigator will pull on. The capability to answer a 483 quickly and credibly when one lands.
That capability has a name on an org chart. A VP or Head of Quality brought in 18 months ahead of a filing does something a Phase 3 readout cannot. The readout gets the science over the line. The quality leader gets the product through the gate that comes after it. In a lot of the programmes we see, that hire is made late, or made junior, and the cost of the decision only becomes visible when the letter arrives.
This is where our work sits. ProGen Search runs retained searches for exactly these roles: Head and VP of Quality, CMC, MSAT and technical operations, and the regulatory affairs leadership that steers the filing itself, across radiopharma, ADCs, cell and gene therapy, and the CDMOs that manufacture for all of them. What the market pays for that calibre of leader is something you can benchmark before you brief a search, and which seats a business at your stage should already have filled is what our Diligence Gap diagnostic is built to expose. We read these letters closely because they are, in the end, a map of where senior manufacturing and quality talent earns its keep.
What We Take From This
The reassuring version of events is that the FDA is broken. It asks nothing of you, because the problem lives somewhere else. Every batch of these letters, the 202, the 89, and now the 14, puts the problem back inside your own walls.
That is better news than it sounds. A readiness problem is one you can fix, if you start early enough and put the right people on it. The molecule is rarely the thing standing between a company and approval. The factory, and the team running it, usually is.
If you are building toward a filing and want a clear-eyed read on whether your manufacturing and quality bench is where it needs to be, that is a conversation worth having early.
Pressure-test your own filing
Two free ProGen diagnostics go straight at the readiness question these letters expose. The FDA Filing Diagnostic pattern-matches your programme against the deficiencies that actually stop applications, so you can see where a filing is most exposed before the agency does. The Diligence Gap shows which senior seats a business at your stage should already have filled, which are exposed against the next milestone, and how long each remaining hire realistically takes to land.
Sources and Notes
This analysis draws on the FDA's public Complete Response Letter database and company disclosures current at the time of writing.
- The 14 letters analysed were released by the FDA in July 2026 and are dated 23 April 2026 (Grace Therapeutics, nimodipine injection, NDA 220304) through 9 July 2026 (Hengrui and Elevar, rivoceranib, NDA 216586). They are the first tranche the agency published after pausing releases in April. Read directly from the letters.
- The deficiency classification is ProGen Search's own, coded from the text of each letter. The FDA frequently redacts the specific deficiencies under exemption (b)(4); the section headings that identify the type of deficiency (product quality, facility inspections, clinical, nonclinical) generally remain visible, and the counts here are based on those. Categories overlap, because most letters cite more than one deficiency.
- An independent read of the same 14 letters by Citeline reached a similar conclusion, with quality and facility issues dominant and only a few letters citing clinical, clinical-pharmacology or statistical deficiencies.
- The named examples are public and company-disclosed. Hengrui and Elevar's liver-cancer combination received its third CRL, on manufacturing and facility-inspection grounds; the combination's second component was refused pending an approval action on its partner drug. Grace Therapeutics' nimodipine injection was refused on product quality alongside a facility inspection, and the company has stated it intends to resubmit.
- The earlier 202 and 89 letters are prior FDA releases. The manufacturing and quality emphasis noted across them reflects ProGen Search's earlier analysis of those batches.
- A CRL is a delay, not a final rejection. Several of these sponsors have said they intend to resubmit.
This article is independent market intelligence and not investment, legal or regulatory advice. It analyses public FDA Complete Response Letters and company disclosures believed reliable at the time of writing. Specific deficiency details in these letters are frequently redacted by the FDA; the classifications here reflect ProGen Search's reading of the visible text. Company names and trademarks are the property of their respective owners. © 2026 ProGen Search Limited.